A data-driven and quantum chemistry-anchored framework for modeling and classifying carbon–lithium bonding in organolithium aggregates
Dec 9, 2025·



Valerii Verkhov
Stepan Meshalkin
Dr. Alexander Antonov
Corresponding author
Prof. Elena Tupikina
Corresponding author
·
0 min read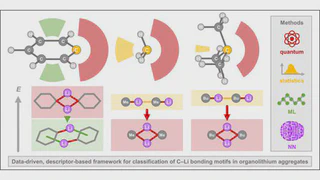
Abstract
We present a quantum chemistry-based, data-driven framework for the automated classification of carbon–lithium bonding motifs in archetypal organolithium aggregates. Starting from ab initio potential energy surfaces-guided sampling, we constructed a chemically complete dataset of 81 optimized gas-phase aggregates of methyllithium, t-butyllithium, and phenyllithium (600 C–Li bonds in total) spanning all relevant nuclearities and bonding modes. Twenty geometric, electronic, and topological descriptors obtained from quantum theory of atoms in molecules and Electron Localization Function (ELF) analyses were evaluated via correlation clustering, yielding a minimal, non-redundant, chemically meaningful set dominated by the ELF basin electron population and key bond-path metrics. This reduced descriptor set was used to train two complementary supervised models—a multi-task fully connected neural network and a bootstrap-aggregated decision tree—achieving accuracies of 84% (nucleophile type), 89% (aggregation state), and 84% (bond type) on validation data. Both methods consistently identified ELF-derived descriptors as the most discriminative, enabling physically grounded separation of bonding regimes (2c–2e, multi-center, non-classical, π–Li) and providing an interpretable, transferable platform for high-throughput bonding analysis in organometallic chemistry.
Publication
J. Chem. Phys. 2025, 163, 224116.



